Journal of
eISSN: 2379-6359


Introduction: The hypogonadotropic hypogonadism associated with disturbances of smell (HHAAO), is a variant of hypogonadotropic hypogonadism, which are associated to a defect in the pituitary or hypothalamus, caused by a lack of hormones which used to stimulate the ovaries or the testicles.
Clinical Case: Based on patients who consulted for disorders of smell, from October 2013 to October 2014, 30 patients aged 6-16years were detected, 3 women under 15years old; they had anosmia that were proven by olfactometry and absence of olfactory bulbs in Nuclear Magnetic Resonance. One patient had lost of hearing too.
Discussion: The study protocol consist of: complete anamnesis, smell test, Nuclear Magnetic Resonance Solid craniofacial smell protocol, hormonal assay of LDH and basal FSH, Serviced inter consultation Pediatrics and Endocrinology. In addition, audiometric studies, abdominal ultrasound and molecular genetics to diagnosis.
LH, luteinizing hormone; FSH, follicle stimulating hormone; GnRH; gonadotropin releasing hormone
The hypogonadotropic hypogonadism associated with disturbances of smell, is a variant of hypogonado tropichypogonadism, these are associated with a defect in the pituitary or hypothalamus. Its origin is due to a lack of hormones that normally stimulate the ovaries or testes: follicle stimulating hormone (FSH) and luteinizing hormone (LH). This is the way how the hypothalamus pituitary gonadal create the axis through which the hypothalamus produce neurons neuropeptide gonadotropin which release (GnRH) stimulates the previous pituitary gonadotrophs more closely the cells to produce the follicle stimulating hormone (FSH) and luteinizing hormone is generated (LH); This stimulate the ovaries and testes to secrete hormones that lead to normal sexual development in the puberty. This axis has its growth in early stages of embryonic development, generating afterwards their functional integration across different neuro endocrine mechanisms of positive and negative feedback necessary to complete secondary sexual characteristics for proper reproductive function. Several alterations in hormone-releasing this axis causes a lack of sex hormones and prevents normal sexual maturation.1 There are multiple causes that can lead to hypogonadotropic hypogonadism and they can be congenital and acquired,2,3 permanent or temporary.4 They can also be divided into general and specific (Male and Female).
In this literature review is mainly addressed hypogonadotropic hypogonadism produced by hypothalamic partial or total disruption.5 Among the causes of hypothalamic hypogonadotropic hypogonadism, the most common are congenital; these are characterized by lack of development of secondary sexual characteristics. This include: defects in migration of GnRH neurons (Kallmann syndrome), alterations in GnRH synthesis (related to the gene encoding the synthesis of GnRH) or pituitary unresponsiveness GnRH in the absence of specific receptors.5
Retrospective analysis of service attended patients before the study.
History
Collect information about relevant clinical data and patient history. Personal and family history regarding the symptom.
Supplementary examinations
In the differential diagnosis of HH we should be present delayed puberty in teenagers, to make a correct diagnosis according to the symptoms presented by the patient.5 It`s really important to work in multidisciplinary teams (Table 1). This work is based on patients evaluated between October 2013 to October 2014 in our department of Otolaryngology, including 3 women under 15years of age, all of them had anosmia proven by olfactometry and absence of olfactory bulbs were detected NMR One patient had hearing loss. Actually some patients are being in study to have an accurate diagnosis of HHAAO, who will be starting their treatment by the specialty to improve their quality of life (Table 2).
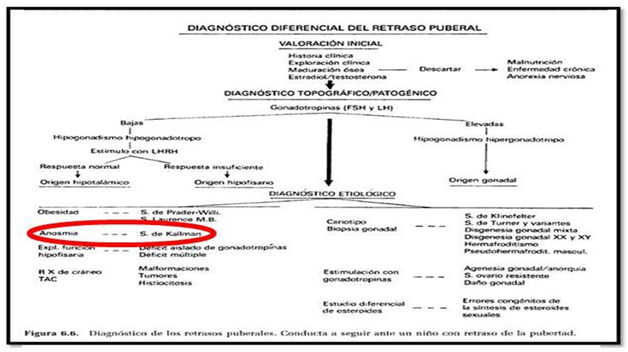
Table 1 Extracted From Hernández Rodríguez. Puberty. Pediatrics
Patient |
|
|
Laboratory Under Basal Conditions |
|
|
|||
Clinic History |
Age |
Olfatometry |
LH (u/l) |
R.M.N |
Abdominal Ultrasound |
Tone Audiometry |
Genetic Examination |
|
Date |
FSH (u/l) Testoterona (ng/dl) |
Protocol Smell |
||||||
Personal Data |
||||||||
Example |
x |
Result |
Result |
Result |
Result |
Result |
Result |
|
Example |
x |
Result |
Result |
Result |
Result |
Result |
Result |
|
Example |
x |
Result |
Result |
Result |
Result |
Result |
Result |
|
Table 2 Protocol proposed work. Dr. Ana R. Cofré. Medical Specialist Otolaryngology GEOG (Study Group Smell and Taste)
The hypogonadotropic hypogonadism Associated Smell alterations or Kallman syndrome was described by Aureliano Maestre de San Juan in 1849 before a picture of hypogonadism-anosmia. Almost a century later Franz Kallman, doctor of German origin psychiatrist based in USA, was one of the pioneers in the study of the genetic basis of these disorders. In 1944, he described a congenital endocrine entity, called hypogonadotropic hypogonadism associated with anosmia, which has become known as Kallmann syndrome.9 Most cases of Kallmann syndrome present with inheritance pattern sporadically but sometimes they are family inheritance. There are three transmission patterns: linked to the X chromosome, autosomal dominant and autosomal recessive,9 the most frequent pattern of X-linked transmission.10A,10B,11 This syndrome affects 1 in 10,000 males and 1 in 50,000 females.12 In males are associated to a preponderance of the pattern of X-linked inheritance.13
In congenital causes there have been identified 5 genes causes: KAL1, FGFR1, PROKR2, PORK2 and FGF8.14 Kallman syndrome or hypogonadotropic hypogonadism associated with disturbances of smell, is also associated in some cases of unilateral hearing lost, can be conductive or sensorineural, usually due to mutations in KAL1, FGFR1, PROKR2 And CHD7 genes.15 Other changes that can be found are renal agenesis, shortening of the metacarpals, reflects synkinesis (uncontrolled muscle movements), cleft palate, dental agenesis, sensorineural hearing loss and seizures. The hypogonadotropic hypogonadism and Kallmann syndrome has a usually late diagnosis in adolescence or adult, usually patients derivatives from ENT evaluation are referrals from other specialities who consulted mostly by problems in fertility. In our case it is a patient, which includes two cases, attended the clinic for suspected abnormalities nose for immediate family and one of the cases that began to notice changes in his nostrils.
An early detection is very important in order to improve the life quality. So the realization of this protocol study of patients at early ages is proposed, trying to include in the initial anamnesis a simple question -"notice changes in your child's nose ...."; should always consider that sense smell forgotten pyramid aristocratic sense, this little giant that opens the doors of an invisible world of sensations.16 Its descent or lack could be considered an early symptom, creating suspicion and orient toward the syndromic diagnosis of hypogonadotropic hypogonadism associated with disturbances of smell, so an early check in life is essential. The purpose of this protocol study was directed by an early detection, reaching a clinical diagnosis and would also be important to perform a molecular diagnosis but this is not done in our country, and it takes very high cost due to the obligation of sending samples abroad.17
Case 1
Patient 9, female, the query was made by his mother because she was worried to notice that her daughter did not feel unpleasant smells then she noticed that she also perceived the basic smells. On otolaryngology exam we detected in the patient that she was the only one in her family with blue eyes with a depigmentation of the iris, what led to research on genetic entities that are associated with patient characteristics. The mother also referred normal pregnancy, normal childbirth, and normal schooling; Childhood diseases: no relevant data.
Audiometry: Left Ear: low perceptual decline in frequencies 4000 and 8000Hz Right ear. Cophosis
Logo audiometry: OD Cophosis, OI Normal.
Olfactometry: Congenital anosmia.
NMR rocks: Normal.
NMR images Protocol 1.5 3D: No formation of olfactory bulbs is evident, or olfactory streak, rudimentary olfactory groove is observed (Figure 1).



Case 2
Patient female 8years old from Entre Rios, her mother noticed disturbances in smell. No relevance clinical history.
Audiometry: Normal.
Olfactometry: Congenital Anosmia.
NMR Protocol smell: No formation of olfactory bulbs is evident or olfactory streak. It is currently being studied for pediatrics (Figure 2).

Figure 1 Case 2
Case 3
15years old female patient who noticed that she couldn`t smell fragances, physical body with features of childishness, referred to the interrogation she does not remember to smell anything before, she does not know chocolate smell for example. No alteration of taste. Irregular menstrual cycle.
Audiometry and logo audiometry: Normal
Olfactometry: Congenital Anosmia.
NMR Protocol smell: No formation of olfactory bulbs is evident or olfactory streak.
Suspecting Kallman syndrome was derived Endocrinology and Gynecology (Figure 3).



The hypogonadotropic hypogonadism is usually diagnosed in adolescence or adulthood. It is important an early to improve the quality of life of this patients. The decrease or lack of smell is an early symptom in hypogonadotropic hypogonadism associated with disturbances of smell therefore we consider that it`s necessary to include an smell test in childhood, at early ages. In front of the suspect and diagnosis the treatment will be in charge of endocrinology and pediatrics, mainly based on hormone replacement therapy to induce puberty and improve fertility.
None.
Author declares there are no conflicts of interest.
None.

© . This is an open access article distributed under the terms of the, which permits unrestricted use, distribution, and build upon your work non-commercially.